
|

| home | contact | press | arabidopsis | yersinia | fowlpox | mpss | cacao |
|
Here as an example we show how GenBank cacao ESTs can be sorted into usable and unusable sequences using the PyMood Sequence Processor. Processing and Masking Cacao ESTs Cacao EST sequences were retrieved from GenBank at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=nucleotide using the following Boolean string: txid3641[Organism] AND EST[PROP] We downloaded the 6557 ESTs into one FASTA file and saved it as a cacao.fasta file. 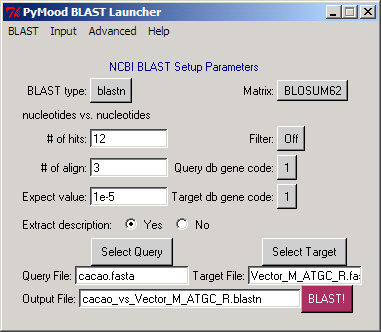 Step 1. Blast for Sequence Processor
Step 1. Blast for Sequence ProcessorDuring this step the query fasta file is compared to a reference fasta file that contains undesired sequences. We selected:
The BLAST run can take from a few minutes to many hours depending on the filesize and the computer processor capacity. 6557 cacao ESTs are typically BLASTed against the supplied reference file "Vector_M_ATGC_R.fasta" in a few minutes. Upon completion of the BLAST run, PyMood BLAST Launcher / Parser produced the following nine files: cacao.annotation Vector_M_ATGC_R.annotation cacao_vs_Vector_M_ATGC_R.blastn cacao_vs_Vector_M_ATGC_R.blastn.matrix cacao_vs_Vector_M_ATGC_R.blastn.matrix.subj.annotation cacao_vs_Vector_M_ATGC_R.blastn.matrix.all_hits cacao_vs_Vector_M_ATGC_R.blastn.matrix.blast_stat cacao_vs_Vector_M_ATGC_R.blastn.matrix.info1 cacao_vs_Vector_M_ATGC_R.blastn.matrix.info2 The detailed description of these types of files is available at our PyMood BLAST Launcher / Parser page 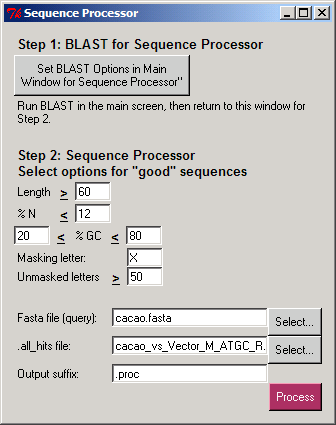 Step 2. Sequence Processor
Step 2. Sequence ProcessorDuring this step the resulting new FASTA files and tab-delimited summary files are produced. Here we made the following selections for cutoffs:
12% as the maximum allowed for N letters 20% to 80% as allowed GC content The last two options affect only ".good.fasta" sequences. We selected:
50 as the minimum required number of unmasked letters in a sequence to be placed in the good.masked.fasta file. (others go in good.maskedx) Here we selected: cacao.fasta as the the query file for processing. cacao_vs_Vector_M_ATGC_R.blastn.matrix.all_hits as the corresponding .all_hits file. The processing of this particular combination took a few seconds. Output Files produced by PyMood Sequence Processor cacao.proc.stat A tab-delimited file with data on sequence composition for each sequence in the original query cacao.fasta file. cacao.proc.all.fasta The original query fasta file formatted so that each sequence is in one line. cacao.proc.bad.fasta A new fasta file containing only sequences that have not passed the first three selected options in the Sequence Processor. cacao.proc.good.fasta A new fasta file containing only sequences that have passed the first three selected options in the Sequence Processor. cacao.proc.good.masked.fasta A new fasta file containing only sequences that have passed all selected options in the Sequence Processor and have the undesired parts masked with the masking letter. cacao.proc.good.maskedx A new fasta file containing sequences that passed the first three selected options in the Sequence Processor but have not passed the last option. cacao.proc.masked.list - a tab-delimited file produced during processing, contains four columns with the information on sequences that have hits to the query fasta file, where the columns are:
A. cacao sequence GI ID
cacao.proc.tab_all a tab-delimited file produced from the cacao.proc.all.fasta fileB. ID(s) of the hit sequence(s) from the target fasta file Vector_M_ATGC_R.fasta C. Length of the alignment D. Length of the cacao sequence cacao.proc.tab_bad a tab-delimited file produced from the cacao.proc.bad.fasta file cacao.proc.tab_good a tab-delimited file produced from the cacao.proc.good.fasta file These three files have three columns:
A. cacao sequence GI ID
B. sequence length C. sequence composition |
|
|