PyMood Sequence Processor
This function removes and masks undesired (low quality, vector, contaminant, specific motifs, etc) sequences from FASTA files. It sorts DNA and protein sequences according to:
- Their level of homology to reference sequence files. The reference FastA file can contain vector sequences, repeats, primers, or any other undesired motifs (nucleotide or proteins) that need to be excluded from subsequential analyses. For our users we supply a fasta file that contains masked vectors (the GenBank collection), all mononucleotide, dinucleotide, and trinucleotide repeats.
- The sequence composition: length, percentage of "N" letters, and "GC" content. The user selects the cutoffs for each option.
 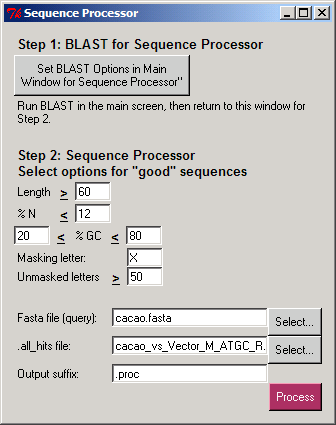
The Sequence Processor separates sequences from a query file into a new ".good" FASTA file (where each sequence has at least some good content) and a ".bad" FASTA file (having no good content). The undesired portions of the ".good" sequences are masked, so they are not used in subsequent analyses. The user selects a letter that will be used to mask the undesired portions of good sequences
and the minimum number of unmasked letters in a sequence to be placed in the ".good.masked.fasta" file.
PyMood Sequence Processor will also generate a number of tab-delimed files containing information on the results of the sequence
processing analysis. They are described in the Cacao example project.
|

