
|

| home | contact | press | arabidopsis | yersinia | fowlpox | mpss | cacao |
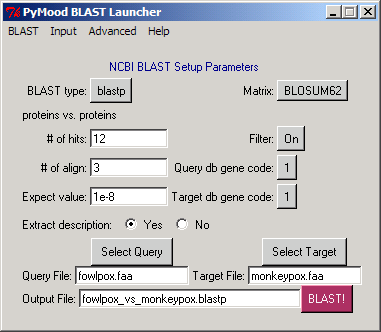 For every comparison, upon completion of BLAST run, PyMood BLAST Launcher / Parser will produce the following nine files : .*blast*The exact extension will correspond to the BLAST type selected (blastn, blastp, blastx, tblastn, or tblastx). An example would be: fowlpox_vs_monkeypox.blastpThese are the text files, and can be opened in a text editor. .annotationFor both the query fasta file and the target fasta file, PyMood BLAST Launcher will produce FileName.annotation files. Examples are:fowlpox.annotation and monkeypox.annotation Each annotation file is a tab-delimited text file, consists of two columns and can be opened in a spreadsheet program or text editor. The first column contains the unique identifier for every gene, and the second column contains the gene name and/or annotation. Annotation in this file is extracted from the corresponding FASTA files. .*blast*.matrix.subj.annotationFor each target fasta file, PyMood BLAST parser will produce a FileName.*blast*.matrix.subj.annotation file. An example is:fowlpox_vs_monkeypox.blastp.matrix.subj.annotation This annotation is extracted from the corresponding .blastp file and contains the annotation data for only those sequences from the target fasta file that are present in the BLAST output file. Each annotation file is a tab-delimited text file, consisting of two columns and can be opened in a spreadsheet program or text editor. The first column contains the unique identifier for every gene, and the second column contains the gene name and/or annotation. .*blast*.matrixFor every BLAST output, PyMood BLAST parser will create a matrix file with the extension .matrixAn example given here is: fowlpox_vs_monkeypox.blastp.matrix This is a tab-delimited text file, and can be opened in a spreadsheet editor. The file contains eight columns, where: A. unique identifier for the query gene B. unique identifier of the best BLAST hit in the target FASTA file, if it is above the cutoff of the Expectation value set in BLAST Launcher C. normalized1 expectation values D. score (bits) E. percentage of identity between the overlapping regions F. number of identical letters in the overlap G. length of the overlap H. shows 1 if there is a BLAST hit found 1 normalized expectation value reflects the absolute value of the exponent in the expectation value. If the expectation value is 2e-15, the normalized expectation value is 15. For all expectation values better than e-100, like 3e-101, 7e-156, etc., the normalized expectation value 100 is assigned. .*blast*.matrix.all_hitsFiles with the extension .all_hits are tab-delimited text files, containing 14 columns with the results from BLAST output, and can be opened in a spreadsheet editor. An example here is:fowlpox_vs_monkeypox.blastp.matrix.all_hits The description of the columns: A. unique identifier for the query gene B. unique identifiers of all BLAST hits in the target fasta file (above the cutoffs set in BLAST Launcher) C. normalized expectation values for all hits D. score (bits) E. percentage of identity between overlapping regions F. number of identical letters in the overlap G. length of the overlap H. order number of the hit for every query gene I. assigns number 1 to every primary alignment1, assigns numbers 2, 3, 4, etc. to the alternative alignments2, if any J. indicates if the alignment is primary, 'PRM' or alternative, 'ALT' K. first position of the query sequence in the alignment L. last position of the query sequence in the alignment M. first position of the target sequence in the alignment N. last position of the target sequence in the alignment 1 the alignment is primary when it is the best scored alignment between the query sequence and the particular target sequence 2 the alignment is alternative when it is not the best scored alignment between the query sequence and the particular target sequence .*blast*.matrix.info1An example here is: fowlpox_vs_monkeypox.blastp.matrix.info1This is a tab-delimited text file, and can be opened in a spreadsheet editor. The file contains eight columns, where: A. unique identifier for the query gene B. unique identifier of the best BLAST hit in the target fasta file, if it is above the cutoff of the Expectation value set in BLAST Launcher C. annotation of the best BLAST hit in the target fasta file D. normalized1 expectation values E. score (bits) F. percentage of identity between the overlapping regions G. number of identical letters in the overlap H. length of the overlap I. shows 1 if there is a BLAST hit found 1 normalized expectation value reflects the absolute value of the exponent in the expectation value. If the expectation value is 2e-15, the normalized expectation value is 15. For all expectation values better than e-100, like 3e-101, 7e-156, etc., the normalized expectation value 100 is assigned. .*blast*.matrix.info2An example here is: fowlpox_vs_monkeypox.blastp.matrix.info2This is a tab-delimited text file, and can be opened in a spreadsheet editor. The file contains 13 columns with the results from BLAST output, where: A. unique identifier for the query gene B. unique identifiers of all BLAST hits in the target fasta file (above the cutoffs set in BLAST Launcher) C. annotation of the BLAST hits D. normalized1 expectation values E. score (bits) F. percentage of identity between overlapping regions G. number of identical letters in the overlap H. length of the overlap I. order number of the hit for every query gene J. first position of the query sequence in the alignment K. last position of the query sequence in the alignment L. first position of the target sequence in the alignment M. last position of the target sequence in the alignment 1 normalized expectation value reflects the absolute value of the exponent in the expectation value. If the expectation value is 2e-15, the normalized expectation value is 15. For all expectation values better than e-100, like 3e-101, 7e-156, etc., the normalized expectation value 100 is assigned. .*blast*.matrix.blast_statAn example here is: fowlpox_vs_monkeypox.blastp.matrix.blast_statThese tab-delimited text files contain a summary of BLAST output statistics. The files consist of three columns, and can be opened in a spreadsheet editor: A. sequence order number B. unique identifier for the query fasta file sequence C. number of primary alignments in the BLAST file for the given query file sequence |
|
|